TRU DEFINITION:
TRU is an acronym for TRansUranic actinides, which are atoms with atomic numbers greater than 92 that are formed when the
abundant uranium isotope U-238 is exposed to a thermal neutron flux.
Typically in used CANDU reactor fuel: the TRU concentration is about 4 grams / kg, about (1 / 2) of the TRU is Pu-239 and about (1 / 6) of the TRU is Pu-240.
APPLICATION:
TRU concentration is of interest to parties
that:
1) Have an inventory of used CANDU reactor fuel and who
would like to use that used fuel to make FNR fuel;
2) Have an inventory of used nuclear fuel from water cooled reactors and who
would like to mitigate the costs of transporting, storing and/or
reprocessing of that used fuel;
3) Have an inventory of used
nuclear fuel from water cooled reactors and who would like to rapidly
convert that used fuel into stable elements that pose no risk to
future human generations;
4) Seek to prevent nuclear weapon
proliferation or further nuclear waste formation via use of closed
system electrolytic fuel reprocessing (pyroprocessing) but who want to minimize the
overall cost.
THE ECONOMIC STRATEGY:
The economic fuel recycling strategy is to minimize the used fuel
reprocessing, shipping and storage costs by performing
relatively simple preliminary used fuel concentration operations on each CANDU power reactor site.
These preliminary
operations include:
a) Chopping up the used fuel into short lengths;
b) Prebaking the chopped fuel to expell and recover vapors and gases (Cs oxides + Kr + Ar +
Xe);
c) Harvesting the used Zr hulls;
d) Dissolving the used fuel in nitric acid;
e) Use of a cascade to divide the used fuel atoms into one large minimally radioactive UO2 portion
(90%) and one smaller highly radioactive portion (UO2 + FP + TRU)(10%) as required for
economic production of FNR fuel;
f) Acid recovery
TRU Concentration:
1) Minimizes the mass of CANDU fuel concentrates
that must be transported to a remote fuel reprocessing site;
2) Reduces the gamma emission from the extracted uranium oxide
sufficiently to make that uranium oxide more economic to transport
and store for future use as the main component of FNR blanket
fuel.
3) Enables complete disposal of TRU from used water cooled
reactor fuel, thus eliminating the necessity of DGRs for nuclear fuel
waste disposal;
4) Minimizes the used fuel mass that must be pyroprocessed;
TRU CONCENTRATION:
TRU concentration is a physical process that concentrates TRU by
selectively extracting nearly pure uranium oxide from used reactor fuel.
This web page describes an apparatus for concentrating the TRU in used
nuclear power reactor fuel about 10X. This apparatus uses a closed
recrystalization process
to separate the used nuclear fuel bundle atoms into three molecular
groups: pure UO2, Zr and everything else (UO2, TRU oxides, fission
products). This TRU concentration process is of fundamental
importance in economic production of fuel for Fast Neutron Reactors
(FNRs).
The largest used fuel portion, which comprises about 90% of the used nuclear fuel
weight, consists of nearly pure uranium oxide. The remaining high purity depleted
uranium oxide has a very low radioactivity and should be stored for
future use as FNR blanket rod material.
The ratios of the
uranium isotopes in this portion are determined by the neutron
irradiation history of the nuclear fuel. For used CANDU fuel this
portion has a very low radioactivity permitting relatively easy and
inexpensive handling, transportation, storage and reprocessing with
minimal gamma ray shielding requirements. Nearly pure UO2 extracted
from used CANDU fuel has a low radioactivity whereas UO2 extracted
from used Light Water Reactor (LWR) fuel has a higher radioactivity
due to the presence of a larger fraction of U-232. The required
amount of UO2 shielding is set by the U-232, U-235 and Np-237
concentrations in the uranium oxide as well as impurities. These
uranium isotopes will be present at low concentrations in the nearly
pure UO2.
The smaller
portion (10%) contains the balance of the used nuclear fuel weight.
For used CANDU fuel this portion is typically: oxides of U, TRU and
fission products. This portion is intensely radioactive and must be
handled and shipped in suitable shielded containers that
have walls that have a gamma ray absorption thickness the equivalent
of a 30 cm thickness of lead and must be stored in dry shielded
containers or vaults.
TRU CONCENTRATION OPERATIONAL OBJECTIVE:
Selective extraction of
uranium oxide should be done at existing CANDU reactor sites to
realize about 90% of the spent CANDU fuel weight as nearly pure
uranium oxide and the remaining 10% consisting of: (about 2.45% of
spent CANDU fuel weight as a mixture of fission products + TRUs) +
(remaining 7.55% of the CANDU fuel weight is uranium oxide).Â
The
uranium content of this mixture is used to meet the uranium metal
content requirement of the FNR fuel. The extracted nearly pure
uranium oxide must be sufficiently pure to reduce its radioactivity
sufficiently to enable low cost transportation and storage.
PROCESS OVERVIEW:
About 2010 Peter Ottensmeyer realized that there is a way of economically converting
used CANDU reactor fuel into FNR fuel with no spurious waste streams.
The method, now termed the Ottensmeyer Plan, has two major steps. The first major step is TRU concentration.
The second major step is pyroprocessing. This web page focuses on TRU Concentration.
TRU CONCENTRATION SUMMARY:
The TRU concentration operations
that need to be performed at each
CANDU power reactor site
include:
a) Chopping up the CANDU fuel bundles to expose the UO2
pellet material;
b) Baking the chopped up used fuel at 650 deg C in a
vacuum oven to drive off and capture Cs, other volatiles and the inert gas fission
products (FP);
c) Dissolving the residue in nitric acid to make
a warm saturated uranyl nitrate hexahydrate [UO2(NO3)2.6H2O]
solution;
d) Separating pure uranium nitrate hexahydrate from
other substances using a 13 stage recrystallization cascade;
e) Nitric acid recovery leaving a 90% pure UO2 pile and a 10% residue
pile.
PROPOSED MECHANICAL USED CANDU FUEL FEED:
a) From the used CANDU fuel inventory that has been
out of a CANDU reactor for at least 10 years withdraw used CANDU fuel
bundles as required.
b) Mechanically shear the used CANDU fuel
bundles into small pieces, each about 3 cm long.
VACUUM BAKING
2) Move the [U3O8 +
contamination Cs2O,Cs2O2, Cs2O3] from #1 above to a suitable furnace.
3) Heat the [U3O8 + contamination Cs2O,Cs2O2, Cs2O3] residue above
650 C to drive off the Cs2O, Cs2O2, Cs2O3 as a condensable gas. 4)
Condense and collect the radioactive Cs2O, Cs2O2, Cs2O3. 5) Send the
radioactive Cs2O, Cs2O2, Cs2O3 to fission product 300 year storage.
After 30 years in storage the radioactivity should be dominated by
Cs-137 (30 year half life) and Cs-135 (3.0 X 10^6 year half life).
Use a segragated store in case this material contains other
contaminants.
The main
reason for prebaking the uranium oxide is to exclude Cs-137 and inert
gases from the downstream dissolver. The
Cs isotope Cs-137 is strongly radioactive and should be trapped
during the prebaking and sent to 300 year storage. Also released
during prebaking are the inert gases krypton, xenon and argon that
were trapped within the used nuclear fuel pellets. Since some of the inert
gas isotopes are radioactive these inert gases must be safely
captured and vented far from a metropolis.
These gases can be
caught in an atmospheric pressure cold trap cooled by liquid
nitrogen. The relevant boiling points are:
|
GAS |
BOILING POINT |
|---|---|
|
Ar |
87.3 K |
|
Kr |
119.1 K |
|
Xe |
165.1 K |
|
CO2 |
194.7 K sublimation |
|
O2 |
90.2 K |
|
N2 |
77.3 K |
The cascade is fed by a closed dissolver. The dissolver has a heating coil and a removable fuel basket to enable removal of undissolved fuel and zirconium hulls. In normal operation the dissolver is always hot at 100 degrees C. The dissolver solution is saturated by maintaining an excess of used CANDU fuel. The dissolver receives HNO3 recovered from the two cascade outputs.
The dissolver has a heating coil and a removable fuel basket to enable removal of undissolved fuel and zirconium hulls. In normal operation the dissolver is always hot at 100 degrees C. The dissolver solution is saturated by maintaining an excess of used CANDU fuel. The dissolver receives HNO3 recovered from the two cascade outputs.DISSOLVER OPERATION:
a) The dissolver is a nitric acid resistant tank
with a removeable top. The dissolver tank contains a nitric acid
resistant basket to allow convenient zirconium hull recovery. An
empty basket is lowered into the dry dissolver tank, the dissolver
tank top is replaced and the dissolver tank is evacuated. This
evacuated air is exhausted to the atmosphere. The evacuation valve is
then closed.
b) Then hot nitric acid temporarily stored in the drain
down tank is pumped into the dissolver.
c) Due to the low overhead
pressure in the dissolver tank nitric acid containing a low UO2(NO3)2
concentration flows from the drain down tank into the dissolver where
it is heated to 100 degrees C. Used CANDU fuel is added to the
dissolver via a feed tube. After some time in the nitric acid at 100
degrees C in the dissolver solution becomes saturated with UO2(NO3)2.
Undissolved zirconium pieces collect in the dissolver's bottom
basket. Radioactive inert gas fission products such as krypton bubble
up through the liquid acid and collect in the sealed space above the
acid.
d) Then a controlled volume of the hot nitric acid solution is
pumped from the dissolver tank into tank T1 via the port at the
bottom of the dissolver. Sufficient nitric acid solution remains in
the dissolver tank over this port to prevent the radioactive inert
gas fission products on top of the dissolver solution from exiting
the dissolver via its bottom port.
e) Eventually when the inert gas
pressure over the acid in the dissolver tank becomes too high or when
the dissolver tank full of Zr hulls the dissolver is cooled to 20
degrees C to reduce the partial pressure of remaining HNO3 gas in the
dissolver head space.
f) The inert gas plus some HNO3 gas in the
dissolver head space are evacuated via a cold trap. The HNO3 vapor is
caught in the cold trap. The radioactive inert gases are sent either
to a high stack or to a pressure tank for later safe release to the
atmosphere at a remote location. Ideally the inert gas should be
stored to allow it to naturally decay. After a suitable decay period
vent the residual inert gas to the atmosphere. Note that radioactive
Kr-81, Kr-85 and Ar-39 must be well mixed with the atmosphere.
g) The
nitric acid in the cold trap is isolated and is recycled back to the
disssolver.
h) When the dissolver is full of Zr hulls the dissolver
is drained into the drain down tank.
i) The dissolver tank top is
removed. The basket containing zirconium pieces is removed from the
dissolver and is air dried. The neutron activated zirconium is
harvested from the dissolver basket for future use as a component of
FNR fuel.
j) Transport the neutron activated zirconium and the CANDU
fuel concentrates to the remote irradiated zirconium and fuel stores.
k) The now empty dissolver tank basket is replaced and the dissolver
batch cycle repeats. Note that the dissolver tank is sufficiently
large that one dissolver batch will serve many temperature cycles.
Note that as the system operates the amount of UO2 in the dissolver
gradually diminishes but the acid liquid level in all the tanks
remains almost constant and the UO2(NO3)2 concentration in the
dissolver solution remains almost constant. From time to time new
used CANDU fuel is added via the feed tube. Â
In the dissolver the irradiated zirconium hulls are captured in a basket for later use in FNR metallic fuel alloy. The neutron activated zirconium hulls were originally used in a CANDU reactor for enclosing the uranium oxide pellets used to fabricate CANDU reactor fuel bundles. Irradiated zirconium can be used in the FNR metallic fuel alloys to prevent the plutonium fraction of FNR core fuel from forming a low melting point Pu-Fe eutectic with the Fe fraction of the fuel tube alloy.
RECRYSTALIZATION CASCADE A 13 stage recrystallization cascade which divides the remaining used CANDU fuel into three portions:
The cascade
consists of a series of nitric acid resistant tanks designated by
Tnx. n = 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 and x = a, b. The detail of the connections
to tank T13a, T14b and T1b differs from the other tanks to enable the
solution discharged by these tanks to properly feed the HNO3 recovery
apparatus.HNO3 RECOVERY
The extracted dissolver solution is heated to
evaporate the nitric acid which is recovered and recycled. A
significant amount of energy is required. The resulting dry residue
is the feedstock for successive operations to make FNR core fuel.
Very strong UO2(NO3)2.nH2O from tank T13a and T1b are transferred to the UO2
/acid recovery units. Heating this very strong solution to recover the
UO2 and the contained acid requires a
substantial amount of energy. The very strong solution in the UO2
/acid recovery unit is mildly heated to drive off the nitric acid
which is recycled.
THE NEPTUNIUM ISSUE:
For chemical reasons the TRU neptunium is not
excluded from UO2 during the uranyl nitrate hexahydrate
recrystallization process. This neptunium stays with the UO2 until ultimately it is fissioned in a FNR.
After TRU concentration, for public
safety reasons, the resulting fuel concentrates should then be
transported in shielded containers to a shared remote fuel
pyroprocessing site. The cost of transporting this used fuel portion is
dominated by the cost of transporting the weight of the required
shielded shipping containers. A major safety concern with respect to
this portion is ensuring that the used fuel concentrates will not go critical if
water penetrates the used fuel concentrates container.
Pyroprocessing involves electrolytic molten salt reprocessing of the residue to reduce the oxides to metals, to separate the concentrate components, to send fission products to 300 year isolated safe dry storage for natural decay and to fabricate suitably alloyed FNR fuel rods.
The fission
products are not waste. The fission products include rare earths that
are in high demand in the electrical industry. After 300 years in dry
storage the fission products will need further chemical processing to
extract the valuable elements.
REFERENCES:
For an overview of the Ottensmeyer Plan please review OTTENSMEYER
PLAN.
For an
overview of nuclear fuel waste processing see the paper:
Radioactive
Waste Partitioning and Transmutation.
Reference:Â Japanese
2002 patent
For an
overview of Uranyl nitrate hexahydrate [UO2(NO3)2.6H2O] solubility
see:
Uranyl nitrate
hexahydrate solubility in nitric acid and its crystallization
selectivity in the presence of nitrate salts
A paper on
decontamination factors actually realized is:
Enhancement of Decontamination Performance of Impurities for Uranyl
Nitrate Hexahydrate
A paper
relevant to the U-232 issue is:Â
Uranium-232 Production In Current Design LWRs
Other relevant papers are:
Actinide Partioning Technologies
and
Extraction Selectivity
UO2(NO3)2 SOLUBIlITY IN NITRIC ACID:
0 deg C = 98 g / 100 g H2O
20 deg C = 122 g / 100 g H2O
100 deg C = 474 g / 100 g H2O
The imporance
of this data is that when a saturated uranyl nitrate hexahydrate
solution at 100 deg C is cooled to 20 degree C the weight of crystals
formed per 100 g H2O will be:
474 g - 122 g = 352 g
The initial
solution weight
474 g + 100 g = 574 g
Thus more than 50% of
the initial warm solution weight forms crystals.
Note that there is little merit in further increasing the weight fraction of crystals because, while in theory that reduces the requred number of stages, it sacrifices crystal face washing by the remaining liquid solution.
Email from
Peter:
Off the top of my head uranium nitrate hexahydrate
dissolves in water up to about 66 %, i.e. it is not solid UNH but
diluted with the 34% water. When it crystallizes out it has a density
as a solid of about 2.8 g/cc whereas the density of the solution is
roughly
2.8 x .66 + 1.0 x .34 or about 2.2.
Therefore I expect
the crystals that form spontaneously to sink.
However, if a cold
surface is introduced into the hot solution, then I would expect the
crystals to form on the cold surface and stick there (depending on
the properties of that surface) until they are scraped off.
Â
RECRYSTALLIZATION
PURIFICATION OPERATING PRINCIPLE:
The dissolver temperature is
kept at 100 degrees C, the dissolver solution is saturated by
maintaining an excess of used CANDU fuel in the dissolver tank.
Assume that a
measured amount of solution from the dissolver tank is transferred from tank T0a
to Tank T1a.
Assume that we start with tank T1a holding a
uniform warm saturated liquid HNO3 solution that contains multiple
solutes. Assume that one solute So (uranyl nitrate hexahydrate) is
dominant. The other non-dominant solutes are S1, S2, etc. From the
perspective of the dominant solute So the fraction of each impurity
Sn is:
(Sn / So).
If the solution is gradually cooled it will
form dominant solute crystals surrounded by liquid solution
containing the lesser solutes. We need the dominant solute crystal
formation rate to be sufficiently small that the resulting dominant
solute crystals are sufficiently large that their discharge from the
tank can be mostly prevented by use of a simple grating or course
filter. Now drain off the cool liquid solution portion in Tank T1a to
Tank T1b. Use a course filter on the cool solution drain line to
prevent the crystals flowing out the drain.
The crystals
are highly regular atomic structures. During dominant solute crystal
formation within a saturated liquid solution mamy of the impurities
tend to be excluded from the dominant solute crystal and hence will
accumulate in the surrounding liquid and on the crystal faces. It is
important to maintain solution agitation sufficient to continuously
wash the impurities off the crystal faces. If, after dominant solute
crystal formation, the surrounding liquid is drained off the excluded
impurity concentration in this drained off liquid is about:
(Sn / Sob)
where Sob is the portion of So in the drained off liquid and
the concentration of impurity Sn in the crystals is close to zero.
Now assume
that it is physically practical to make the weight of dominant solute
crystals about equal to the weight of drained off liquid at 20 degrees C. Then:
Sob ~ (So / 2)
The impurity
concentration in the drained off liquid is:
[Sn / Sob]
= (Sn / So)(So / Sob)
~ 2 (Sn / So)
Now warm up the crystals remaining in tank T1a. They are nearly pure with respect to exclusion of impurity Sn. Drain out this liquid to another tank T2a via a micron filter. This filtrate is very pure. The purpose of the micron filter is to try to trap in the tank T1a impurities that were incorporated into the solid crystal but not chemically bound. These impurities will tend to drain out to tank T1b via the course filter at the next opportunity.
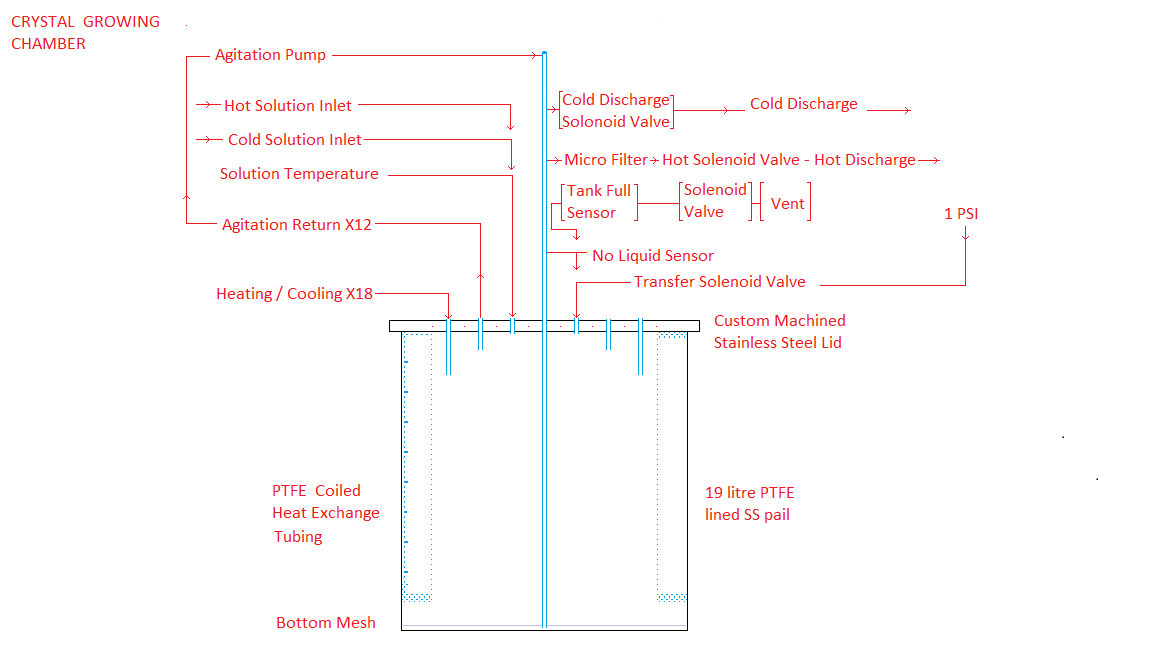
A detailed
description of the design and operation of a typical Tank Tna containing the Uranyl Nitrate
Hexahydrate automatic crystal growing chamber is set out at:
Uranyl
Nitrate Hexahydrate Crystal Growth
RECRYSTALLIZATION PROCESS CONSTRAINTS:
1) The process equipment must not be so
large that a critical mass can accumulate anywhere in the apparatus.
We must be careful with respeect to tank T1b where the impurity concentration is
highest.
2) Nitric acid (4.5 M) acting on uranium oxide produces
uranyl nitrate hexahydrate. The process of recrystalization of uranyl
nitrate hexahydrate [UO2(NO3)2.6H2O] is used to selectively extract
nearly pure UO2 from spent CANDU fuel. This process is ineffective at
rejecting the elements Np and Cs. The Np simply stays mixed with the
uranium. It is not a radioactivity problem unless it contains the
isotope Np-237. Np-237 arises from fast neutron n > 2n reactions
in U-238. In CANDU reactors the fast neutron flux is very low, so the
Np-237 production is low so the contribution of Np-237 to spent fuel
radioactivity is very low. This statement is not true for spent fuel
from fast neutron reactors. Hence recrystalization of
[UO2(NO3)2.6H2O] will not achieve comparable radioactivity reduction
in FNR spent fuel.
3) The process of
recrystalization of [UO2(NO3)2.6H2O] is also ineffective at rejection
of CsNO3.2H2O from the crystals. Before the recrystalization steps Cs
is rejected by baking the uranium oxide at over 650 degrees C at
which point the isotopes Cs-133, Cs-135 and Cs-137 vaporize as the
oxides Cs2O, Cs2O2, Cs2O3 leaving behind U3O8 with some contaminant
Np. These vapors must be condensed in a cold trap. As indicated above
the contaminant Np is not a problem unless it contains Np-237 from
fast neutrons interacting with U-238.
4) During the initial
prebaking most of the trapped radioactive Kr-81, Kr-85 and Ar-39
inert gas atoms are released in the vacuum furnace. This trapped
inert gas mixture must be captured, condensed, stored, transported,
separated from Cs, adequately mixed with the atmosphere and safely
vented.
5) There must be a mechanism to safely prevent
uncontrolled release of any remaining inert gases or Cs while new UO2
is being added to the dissolver tank.
CHEMISTRY:
U3O8
= UO2 + 2 UO3
UO2 + 2 HNO3 + 6 H2O = UO2(NO3)2.6H2O + H2 + heat
UO3 + 2 HNO3 + 5 H2O = UO2(NO3)2.6H2O
UO2(NO3)2.6H2O + heat = UO3 + 2 HNO3 + 5 H2O
DEFINITIONS:
The
term weak solution refers to a UO2(NO3)2 4.5 M nitric
acid solution which is saturated at 20 degrees C. The term strong
solution refers to a UO2(NO3)2 4.5 M nitric acid solution which
is saturated at 100 degrees C.
HYDRATION OF UO2(NO3)2:
According to Wiki UO2(NO3)2.nH2O exists as a
dihydrate, a trihydrate and a hexahydrate. It appears that unless
concentrated nitric acid is used the result will be the hexahydrate.
However, when the hexahydrate is gradually heated during UO2 recovery
the hexahydrate may liberate water molecules to form the trihydrate
and then dihydrate forms.
The components and design of this crystal growing chamber are described at:
Uranyl Nitrate Hexahydrate Crystal Growth
Typically in
real life a single stage of separation gives:
A ~ 0.95
and
B
~ 0.05
Recrystalization separation factor = (A / B) ~ 19
As (I / U) rises the rejection of impurities on crystal formation becomes less ideal. Impurities that chemically bond with the uranium nitrate hexahydrate are not rejected.
RECRYSTALIZATION CASCADE DESCRIPTION:
The main
apparatus used for selective uranium oxide extraction is a nitric
acid dissolver followed by a thirteen stage uranyl nitrate hexahydrate
recrystallization tank cascade.
The mixed input to the cascade is
(U + I) = uranyl nitrate hexahydrate solution
Im =
impurities (fission products and TRU)
CASCADE OVERVIEW:
In operation hot strong acid solution saturated with
UO2(NO3)2 at 100 degrees C is fed from the dissolver into tank T1a
and cool clean acid solution saturated with UO2(NO3)2.6H2O at 20
degrees C is later generated in tank T13a. As the dissolved used CANDU
fuel moves from tank column 1 to tank column 13 it converts from being
97.55% UO2(NO3)2.6H2O to being nearly pure UO2(NO3)2.6H2O. The
initial impurity fraction in used CANDU fuel is:
2.45% impurity to about 24.5% impurity.
The equipment
consists of a dissolver, tanks Tnx where n = 1,2,3,4,5,6,7, 8, 9, 10, 11, 12, 13, 14 and x =
a,b and two UO2 / acid recovery units. The dissolver contains
100 degree C saturated solution with an excess of used CANDU fuel. The
system goes through a series of temperature oscillations each
consisting of a slow cooling period from 100 degrees C down to 20
degrees C (320 minutes) followed by a more rapid heating period from
20 degrees C back up to 100 degrees C (80 minutes). After each
heating period clean hot solution is transferred one tank to the
right. During the subsequent cooling and crystal growth period TRUs +
FP are rejected from the crystals to the liquid. At the end of each
cooling period the remaining liquid solution flows down to tank Tnb
carrying the rejected impurities toward the impurity output. We can
refer to each complete module temperature oscillation as a
temperature cycle. Due to cleaner solution feedback the solution fed
back to tank Tna is cleaner than the solution that flowed into Tank
Tnb.
CASCADE
DESIGN:
The cascade is designed so that the Tna row tanks
operate at progressively higher purities going to the right and the Tnb row tanks operate at higher
impuity concentrations going to the left. At the end of each heating
period solution is transferred one tank to the right sequentially starting with T13a. While liquid transfers are occurring the liquid volume
in the individual tanks changes.
The tank temperatures are
programmed to oscillate, typically between 20 degrees C and 100
degrees C. The circulated heating oil reaches up to 120 degrees C for
warming and reaches down to 15 degrees C for cooling.
The heating can be relatively fast (~ 1.0 degree C /
minute) but the cooling must be relatively slow (~ 0.25 deg C / minute). The required fine temperature control is
achieved by controlling the circulated oil temperature. During a tank
cooling periood UO2(NO3)2.6H2O crystals tend to grow on the textured
bottom surface but not onthe teflon surfaces.
INTERTANK LIQUID TRANSFERS:
Intertank liquid transfers are realized by
applying one psi air pressure over the upstream tank while venting the
downstream tank. There are two types of intertank liquid transfers.
One type of intertank transfer stops the liquid transfer when the upstream tank indicates that it contains no more liquid. This type of liquid transfer is used when there are solid crystals remaining in the upstream tank. In this case a liquid sensor is mounted on the common liquid discharge tube. When thiss sensor detects no liquid the liquid transfer is complete. Such a liquid transfer from tank T1a to tank T2a is indicated by: Transfer T1a, T2a, NL (No Liquid).
The other type of liquid transfer stops the transfer when the downstream tank indicates that it is full. This type of transfer is used when the upstream tank is intentionally not fully emptied. The downstream tank full sensor is mounted on its vent tube upstream of the vent solenoid valve. Such a transfer from tank T3b to tank T3a is indicated by Transfer T3b, T3a, DF (downstream full).
TANK T1a TO TANK T2a SOLUTION TRANSFER SUBROUTINE, UPSTREAM NO LIQUID DETECTION:
A liquid transfer from
tank T1a to tank T2a is achieved by:
a) Stop solution agitation in
tank T1a;
b) Open the NO top vent valve on the top of tank T2a;
c) Open the NC warm liquid discharge valve branch off the discharge
tube of tank T1a;
d) Close the NO top vent valve on tank T1a;
e) Open the NC transfer air pressure valve on the top of tank T1a;
f) Wait for tank T1a no liquid sensor to indicate empty for one full second;
g) Close the NC transfer air
pressure valve on the top of tank T1a;
h) Open the air vent valve on tank T1a;
i) Close the NC warm liquid discharge valve branch off
the common teflon discharge tube of tank T1a;
TANK T3b TO TANK T3a SOLUTION TRANSFER SUBROUTINE, DOWNSTREAM TANK FULL DETECTION:
A liquid transfer from
tank T3b to tank T3a is achieved by:
a) Open the NO vent valve on the top of tank T3a;
b) Open desired NC liquid discharge route valve branch off the main discharge
tube of tank T3b;
c) Close the NO vent valve on tank T3b;
d) Open the NC transfer air pressure valve on the top of tank T3b;
e) Wait for tank T3a to indicate full for 0.2 seconds;
f) Close the NC transfer air
pressure valve on the top of tank T3b;
g) Open the air vent valve on tank T3b;
h) Close the NC main liquid discharge valve off
of tank T3b;
CASCADE FORMATION:
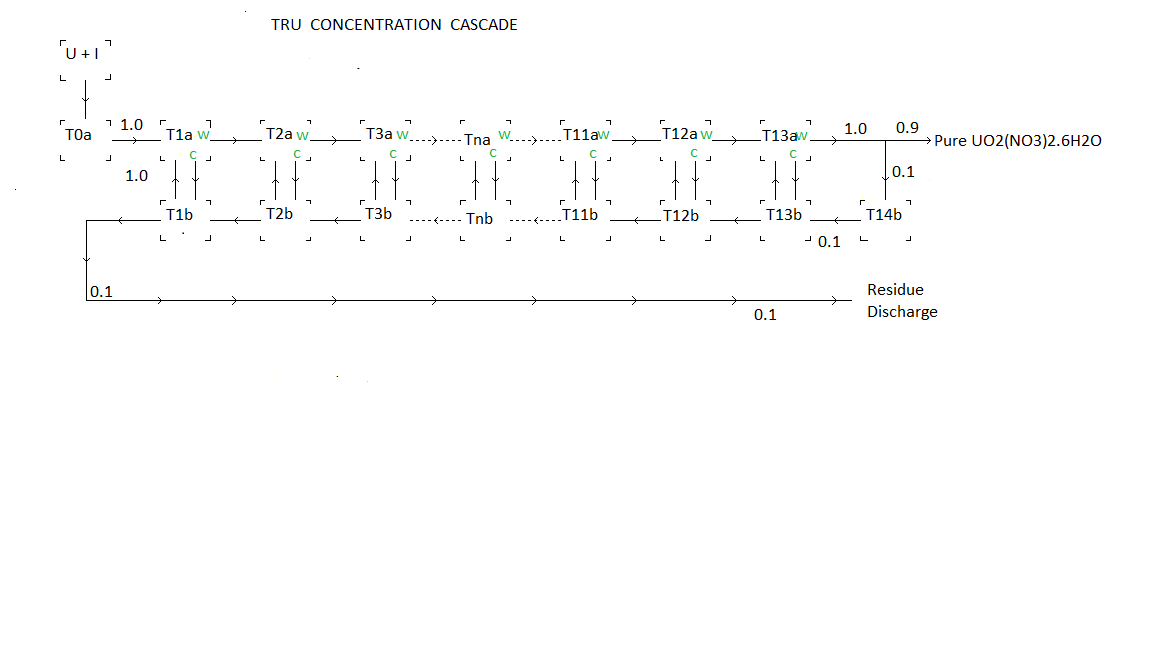
The cascade is formed by sequentially connecting thirteen crystal growing stages with residue feedback.
The cascade operates by taking advantage of the temperature dependence of UO2(NO3)2 solubility in 4.5 M HNO3. Lowering the temperature of a saturated solution triggers recrystallization. During slow recrystallization impurity atoms are excluded from the UO2(NO3)2.6H2O crystals and concentrate in the surrounding liquid solution. In order to realize high purity UO2(NO3)2.6H2O a cascade containing thirteen successive recrystalization steps is used. At low temperatures about half of the solution forms solid crystals. At high temperatures the solution is saturated but there are no solid crystals.
The tanks Tna all oscillate in temperature together. During a Tna cooling period crystals form in the tanks Tna. During the cooling period part of the solution in tank Tnb is shifted into tank T(n-1)b. At the end of a cooling period liquid solution remaining in Tank Tna is transferred into tank Tnb. During the subsequent warming period the crystals remaining in in tank Tna melt. At the end of the warming period this pure solution is transferred right into tank T(n+1)a. Then part of the solution in tank Tnb is shifted into tank Tna.
During steady state operation the fraction:
(TRU + FP) / U at the cascade residue discharge builds up to be about
10X the (TRU + FP) / U fraction in the dissolver, which
fraction is three orders of magnitude higher than the corresponding
value of (TRU + FP) / U at the nearly pure uranium oxide discharge.
CASCADE OPERATION: Tanks
Tna, which each accommodate a solution volume of at least 2 U, oscillate in temperature. During a cooling period crystals form in tanks Tna. During a warming period these crystals melt. The normal operation initial condition is that when full each of tanks Tna contain 2 U of hot saturated solution and each of tanks Tnb contain (U / 10). During the period during which tanks Tna are cooling solution amount (U / 10) is shifted left one position in tanks Tnb starting with T1b and moving to the right to T14b so that at the end of this solution shift sequence tank T14b is empty. Tank T1b discharges (U / 10) to the cascade residue output. The amount (U / 10) is indicated by total discharge of each upstream tank into the next downstream tank as indicated by no liquid in the liquid transfer circuit. The amount U / 10 is set by a measured amount of solution that enters tank T14b which amount is set by the T14b full indicator. Wait until all tanks Tna reach the desired low temperature (20 deg C). At the end of the cooling period all of
the remaining cool liquid in each tank Tna (approximate volume U in each tank) is transferred to the accompanying tank Tnb. Note that tanks Tnb must each accommodate a volume of more than 1.1 U. This transfer occurs until each upstream tank Tna is dry as indicated by no liquid in the common liquid transfer circuit. Then tanks Tna and Tnb are both warmed. During a warming period the solid crystals in tanks Tna melt. Wait until all tanks reach the desired high temperature (100 deg C). Then moving from right to left the warm saturated solution of volume U is completely discharged to the next a tank T(n+1)a as indicated by no liquid in the liquid transfer circuit. The volume (U /10) of the liquid discharged from the last tank T13a is discharged to tank T14b as indicated by a liquid level sensor in the T14b vent line and the then the remaining (9 U /10) is discharged to the
UO2(NO3)2.6H2O cascade output. Volumed U of new saturated solution flows from tank T0a into tank T1a. In each case no liquid in the common liquid transfer line indicates complete transfer. The volume in Toa is indicated by a liquid level sensor in its vent line. Then the warm liquid in tanks Tnb for n = 1 to 13 is transferred into tanks Tna where it is mixed with the newly shifted in liquid
from tanks T(n-1)a so that tanks Tna all contain 2 U of solution. Completion of this liquid transfer is indicated by tank full sensors in the vent lines of tanks Tna. Note that the liquid insertion amount U flowing from tank T0a and recirculated amount (U / 10) flowing from tank T14b are accurately calibrated. SOLUTION SHIFTING SUMMARY: About 90% of the solution resulting from melting
the remaining crystals in tank T13a is transferred into the pure UO2(NO3)2.6H2O output and then the UO2 / HNO3
recovery.
While a tank Tna is being heated or cooled there is
no solution flow in or out of tank Tna. During the cooling period crystals
form which increase the residue concentration in the liquid
surrounding the crystals. At the end of the cooling period crystals
containing nearly pure uranium nitrate hexahydrate are retained by a
screen and the surrounding cool liquid is transferred to tank Tnb.
Then tank Tna is heated. The
UO2(NO3)2.6H2O crystals remaining in tanks Tna melt.
After a warming period, starting from the right, there are successive
warm liquid discharges from tank Tna to tank T(n+1)a toward the nearly pure uranium discharge.
Then starting from the right tank Tna is reloaded from tank T(n-1)a. Then the tank Tna is
reloaded from tank Tnb. Then another cooling period
commences. During this period solution volume (U / 10) is left shifted out of the Tnb tanks starting from the left.
CASCADE MATHEMATICS:
A practical
cascade design is one where recrystallization at each stage is
assumed to be imperfect. Assume that the weight of crystals = weight of solutution at the lowest temperature. Then the single stage transfer function is:
U + I = [(U / 2) + A I] + [(U / 2) + B I]
where cool output flow per thermal cycle is:
[(U / 2) + A I]
and where warm output flow per thermal cycle is:
[(U / 2) + B I]
where:
A + B = 1
U = total uranium nitrate hexahydrate stage input
I = total impurity stage input
CASCADE BOUNDARY CONDITIONS:
Cascade input = (U + I)
Cascade main discharge = (9 / 10)(U) + k I
k < (1 / 1000)
Cascade residue discharge = (U / 10) + (1 - k) I
Ideally with
an infinite number of stages k = 0 so the residue discharge flow
is:
(U / 10) + I
For each Tnb stage for n = 1 to 13:
Main output = 10 X Aux output
For tank T1b:
Main output = 10 [(1 / 10)(U) + (1 - k)(I)]
= U + 10 (1 - k) I
Cascade Separation Factor:
[(I / U)] / [k (I /U)]
= [1 / k]
FLOWS
FIRST STAGE ANALYSIS:
Tank T1a Main input = (U + I)
Tank T1a Aux input = 10[Residue discharge]
= 10 [(1 / 10)(U) + (1 - k)(I)]
= (U) + (1 - k) (10 I)
T1a total input = 2 U + (11 I) - k (10 I)
T1a warm output = (U) + B I[(11) - 10 k]
T1a cool output = (U) + A I [(11) - 10 k]
Tank T1b Main input = (U) + A I [(11) - 10 k]
Tank T1b Aux input = (U / 10) + K2 I
Tank T1b Total input = (11 U / 10) + A I [(11) - 10 k] + K2 I
Tank T1b Aux input = (Tank T1b total input - Tank T1b main input)
= (11 U / 10) + A I [(11 ) -10 k] + K2 I - [(U) + A I [(11) - 10 k]]
= (U / 10) + K2 I
K2 ~ (1)
Tank T1b Total input = Tank Tib total output
or
(11 U / 10) + A I [(11) - 10 k] + K2 I
= (11 U / 10) + (1 - k) 11 I
or
A I [(11) - 10 k] + K2 I = ( 1 - k) 11 I
or
A [(11) - 10 k] + K2 = (1 - k) (11)
or
K2 = (1 - k)(11) - A [(11) - 10 k]
= 11 (1 - k - A) + 10 k A
= 11 (B - k) + 10 k A
= 11 B - 11 k + 10 k A
= 11 B - k - 10 k + 10 k A
= 11 B - k - 10 k (1 - A)
= 11 B - k - 10 k B
For the
special case of many stages then:
k = 0:
giving:
K2 I = (11) I (1- A)
= (11 / 2) I B
which, as expected, is the same as the impurity carried forward via the T1a warm discharge.
The effective first stage separation factor is:
(I / U) / [B I ((11) - 10 k) / (U)]
= 1 / [B (11 - 10 k)]
which for k = 0 becomes:
1 / [B (11))]
= 1 / 11 B
which for B ~ (1 / 20) becomes:
20 / 11
LAST STAGE ANALYSIS:
Tank Ta Main Input = Tank Ta Total Input - Tank Ta Aux Input
= Tank Ta Total Output - Tank Ta Aux Input
= 2 U + I - [U + (10 / 11) A I + (B I/ 11)]
= U + I - (10 / 11) A I - (B I/ 11)]
Tank Ta Aux Input = Tank Tb main output
= U + (10 / 11) A I + (B I/ 11)
Tank Ta warm output = U + B I
Tank Ta cool output = U + A I
Tank Ta total output = 2 U = Tank Ta total input
Tank Tb Main Input = U + A I
Tank Tb Aux input = (U + B I) / 10
Tank Tb total input = (11 / 10) U + A I + (B I / 10)
Tank Tb Main Output = U + (10 / 11)[A I + (B I / 10)]
= U + (10 / 11) A I + (B I/ 11)
Tank Tb Aux output = (U / 10) + (A I / 11) + (B I / 110)
Last stage separation factor:
[I - (10 / 11) A I - (B I / 11)] / (B I)
= (1 / B) - (10 / 11)(A / B) - (1 / 11)
= (1 / B) - (10 / 11)((1 - B) / B) - (1 / 11)
= (1 / 11 B) + (9 / 11)
For B = (1 / 20):
Last stage separation factor = (29 / 11)
Thus about 13 stages, each with two tanks, will likely be required to reduce the
Pu impurity concentration in the UO2 by a factor of 1000.
FIRST STAGE
OVERVIEW: The contents
of Tank T1b are heated to 100 degrees C and 1 U of this liquid is recycled back into tank T1a. Then 1 U of the contents
of tank T0a are transferred into tank T1a.
Tank T1a then
starts a new cooling cycle.
The full cascade converts 90% of the from being 97.55%
UO2(NO3)2.6H2O to being nearly pure UO2(NO3)2.6H2O. The initial
impurity fraction in used CANDU fuel is: 2.45%. The impurity
fraction at the cascade residue discharge is ~ 24.5%.
The cascade
consists of a series of nitric acid resistant tanks designated by
Tnx. n = 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 and x = a, b. The detail of the connections
to tank T13a, T14b and T1b differs from the other tanks to enable the
solution discharged by these tanks to properly feed the HNO3 recovery
apparatus.
The tank
discharge tubes are fed from the very bottom of each tank. The
discharge connections to the adjacent downstream tanks are to the
space above the liquid levels in the downstream tanks to ensure that
there are liquid breaks in the connections between adjacent tanks.
Each tank's overhead gas space is vented via a liquid detector and a NO solenoid valve to a common overhead vent
pipe. This solenoid valve is sometimes closed to permit easy liquid transfer
between tanks while safely containing hot HNO3 and related gases.
The tanks Tna
are all weakly agitated during crystal growth by circulating solution so
that there is rising solution convection within the UO2(NO3)2.6H2O
solution. The purpose of the weak agitation is to minimize impurity
accumulation on the crystal faces during crystal growth and to
prevent common drain blockage by small crystals.
Each tank has a
temperature sensor which is used to monitor the heating/cooling
rates and to indicate when a heating or cooling cycle is complete.
During the
heating period the temperature of the circulated oil is about 20 degrees C
warmer than the temperature of the solution. During a cooling
period the temperature of the circulated oil is about 5 degees C cooler than the
solution.
At the end of
the heating and cooling periods the solution in each tank is
transferrred into the next downstream tank.
Absent U-232
in the used fuel this cascade should reduce the uranium oxide gamma
emission per kg down to the level of new CANDU fuel formed from
natural uranium_______??. The amount of U-232 gamma emission will be
a strong function of the original Th-232 impurity concentration in
CANDU fuel and a weak function of the age of the used CANDU fuel. During an
intertank solution transfer each transfer normally runs until either the downstream tank is full or there
is no liquid in the upstream tank.
The tanks all
have liquid level full sensors and common drain tube empty sensors.
There is
control logic which will stop the liquid transfer sequence and alarm
if any tank liquid level exceeds its design maximum. The floor under
the tanks is covered with an acid resistant stainless steel sheet which
is sloped to a common acid drain. The drain goes to a basement level
dirty acid drain down tank. Thus any acid leak anywhere in the system
gravity drains into this dirty acid drain down tank.
CASCADE MAINTENANCE:
The cascade
requires drain and rinse valves at each end to enable cascade draining and
flushing. UO2(NO3)2 SOLUBIlITY: The amount of CANDU fuel processed per thermal cycle
is: Mass per tank per 100 g
H2O that must be heated and cooled in each thermal cycle is: TANK VOLUMES: HEAT REQUIRED TO DRIVE ONE THERMAL CYCLE OF OPERATION: Heat of formation of UO2(NO3)2.6H2O: Heat capacity of H2O = Heat capacity of UO2 = Heat capacity of HNO3 = Latent heat of vaporization of water = Volume of each tank =
In operation hot strong acid solution saturated with
UO2(NO3)2.6H2O at 100 degrees C is fed from the dissolver into tank
T1a main inlet. Tank T1a is then cooled to 20 degrees C. The
remaining cool liquid is transferred from tank T1a to tank T1b.
The remaining solid UO2(NO3)2.6H2O in tank T1a is then warmed. After crystal melting the nearly pure liquid is shifted into tank T2a.
To enable system maintenance the dissolver basket
containing remaining fuel and zirconium hulls is removed and placed
behind a shielded barrier, all the tanks are heated to 100 degrees C
to fully dissolve the remaining solids and then the entire system
solution volume is shifted and drained down into the shielded below grade acid
drain down tanks. Below the drain down tanks are pumps which can be
used to transfer the acid solution back into the cascade after the
service work is complete.
0 deg C = 98 g / 100 g H2O
20 deg C = 122 g / 100 g H2O
100 deg C = 474 g / 100 g H2O
Solute mass
per 100 g H2O that moves one tank forward with each thermal cycle:
(474 g - 122 g) / 100 g = 352 g UO2(NO3)2 / 100 g H2O
[UO2 / UO2(NO3)2] X 108 g / 100 g H2O
= [(238 + 32) / (238 + 32 + 2 (14 + 48))] X 108 g / 100 g H2O
= [(270) / (270 + 124)] X 108 g / 100 g H2O
= 74.01 g / 100 g H2O
100 g H2O + 474 g UO2(NO3)2
The volume of a
basement drain down tank sufficient to absorb the entire volume in
the dissolver, Tnx is: ________ We may need to be concerned about
accumulating a critical mass in the dissolver. Hence, we may need to
rethink these tank sizes. Note that the submerged tube surface area
must be consistent with the assumed heat transfer rate.
The TRU Concentration process has two major requirements for heat.
a) On each thermal cycle the contents of 14 tanks have to be raised from 20 degrees C to 100 degrees C;
b) On each thermal cycle the water content of one tank must be evaporated as part of the acid recovery process.
= -2739.5 Btu/lb of UO2(NO3)2
This is heat that must be provided to drive off the water. ??????
Hence: (1 Btu / lb) = 252.2 cal / 454 g = 0.5555 cal / g
Thus the heat required to recover UO2 from UO2(NO3)2.6H2O is:
2739.5 Btu/lb of UO2(NO3)2 = 2739.5 Btu/lb X (0.5555 cal / g) / (1 Btu / lb)
= 1522 cal / g of UO2(NO3)2.6H2O
Acid Recovery:
Each thermal cycle produces 108 g of UO2(NO3)2.6H2O. Thus the heat
required for UO2 recovery per thermal cycle is:
108 g X 1522 cal / g
= 164,376 cal
= 164,376 cal X 4.18 J / cal = 687,092 J
= 687,092 J X 1 W-s / J X 1 kWt / 1000 W X 1 h / 3600 s
= 0.1908 kWht
Thus the energy required per thermal cycle / 100 g H2O / tank is:
0.1908 kWht + 0.2666 kWht
= 0.45746 kWht / 100 g H2O / tank From above, each thermal cycle produces:
74.01 g UO2 / 100 g H2O
Thus at a facility processing 5000 kg / day of used CANDU
fuel has an average thermal power consumption of:
5000 kg / day X 1 day / 24 h X 0.47546 kWht/74.01 g X 1000 g / kg
= 1338.38 kWt
By the time fan and pump loads for heat removal are added and the
heat capacity of the tanks is included this will be about 2.0 MWt.
Note that this is an average heating power. In reality the system
heats for 80 minutes followed by a cooling period of 320 minutes.
Thus during the heating periods the peak power is 5X the average or:
5 X 2 MWt = 10 MWt.
Note that the UO2 / HNO3 recovery
unit can be run continuously. Hence about 800 kWt is needed
continuously and about 6 MWt are needed with a 20% duty cycle.
SOLVE FOR ACTUAL HEATING AND COOLING TIMES TANK REQUIREMENT:
Each
temperature cycle = 400 minutes. Number of temperature cycles per day
= [24 h X 60 m / h] / [400 min / cycle]
= 3.6 cycles / day
In order to produce 5000 kg / day of UO2, each cycle must produce:
5000 kg / 3.6 = 1389 kg UO2
Each 100 g of acid produces: 74.01 g UO2. Hence the
required number of 100 g units of acid per tank is:
1389 kg / 74.01 g = 18.77 X 10^3
Hence the required amount of acid / tank is:
18.77 X
10^3 X 100 g = 1877 kg
MISCELLANEOUS CASCADE DESIGN ISSUES:
Provide perforated sheet disks
in each tank to provide crystal growth surfaces and to prevent
crystals being sucked into the inlet of a backward pump. These must
be perforated all the way to their bottom edges to ensure complete
drainage. 5) It is necessary to carefully control the liquid
transfers to realize the optimum acid volume in each tank at a
particular time in the operating cycle. If there is too much acid in
a tank the system will be energy inefficient on thermal cycling and
the forward solute propagation will be poor. If there is too little
acid in a tank not all the crystals will be dissolved during a tank
heating cycle, leading to insufficient crystal growth in the next
tank during its cooling cycle. Thus the liquid levels must be
carefully controlled. Each tank should have a precise liquid level
sensing device and a pressure sensor at the tank discharge. The level
control in tank T1 is particularly important as it sets the levels in
the other tanks if the tank flows are all properly balanced. Thus the
lids must have provisions for the required
liquid level sensors. Â
SYSTEM SERVICE:
The envisaged cascade is orders of magnitude more
mechanically simple than the earlier student design summarized below.
From time to time the system will likely need mechanical service. To
safely enable such service both the HNO3 and the radioactive species
must be completely drained. To service the system heat the entire
system to 100 degrees C to dissolve all of the UO2(NO3)2. Lift out
the dissolver basket with a gantry crane and place it in a shielded
enclosure and then drain all the fluids from the drain valves below
the bottoms of the tanks to shielded below grade drain down tanks. If
the radiation from residue remaining in the system is too high flush
the system with clean 4.5 M nitric acid. Â
UO2(NO3)2 DATA:
MP = 60.2 deg C
Dissociation at 118 deg C
UO2(NO3)2 is hygroscopic forming 6(H2O), 3(H2O), 2(H2O)
TOXICITY OF UO2(NO3)2: 12 mg / kg (dog)
Natural Uranium:
U-238 99.27%, 4.47 X 10^9 Y
U-235 0.711%. 700 X 10^6 Y
U-234 ~ 0.019%
Price of uranium oxide ~ $28 / lb
PROCESS ECONOMICS:
Assume that we can process 7,900 ml of solution
per temperature cycle. That is a solution mass of:
(2.2 kg / 1000 ml) X 7,900 ml / cycle = 17.38 kg / cycle.
The weight of uranyl
nitrate hexahydrate UO2(NO3)2.6H2O processed per cycle is:
[(2.8 X 0.66) / 2.2] X 17.38 kg / cycle = 14.6 kg / cycle
Assume 6 temperature cycles per day:
5 cycles / day X 14.6 kg / cycle = 73 kg / day uranyl nitrate hexahydrate
Weight Fraction UO2 = UO2 / [UO2(NO3)2.6H2O]
= (238 + 32) / [238 + 32 + 2(62) + 6(18)]
= 270 / [270 + 124 + 108]
= 270 / 502
= 0.5378
Hence UO2 throughput = 73 kg /day X 0.5378
= 39.25 kg /day
With careful setup adjustment one TRU concentration system will process UO2 at 39.25 kg / day
yielding a TRU output of
39.25 kg / day X 4 g / kg = 157 g / day TRU.
Wiki indicates that The ultimate value of fissile is $7600 / kg
Thus the gross
value of this fissile fuel after pyroprocessing is:
0.157 kg/ day X $7600 / kg = $1193.42 / day
which must include the cost of
pyroprocessing.
Assume that the TRU concentration apparatus costs $500,000 and needs to earn a 2 year simple payback on capital plus $100,000 / year for cost of operation.
Gross income
is $1193.42 / day X 365 days / year = $435,598 year. Hence,TRU
concentration costs $350,000 / year leaving:
435,598 - 350,000 =
$85,598 / year
for pyroprocessing 157 gm / day TRU or .785 kg /
day of FNR core fuel.
One CANDU
reactor produces:
( 4 g TRU / kg UO2) X 100,000 kg UO2 / 1.5 years = 267
kg TRU / year
One TRU
concentrator produces:
0.157 kg TRU / day X 365 day / year = 57.3
kg / year
Thus we need:
267 / 57.3 = 4.66 TRU concentrators at $500,000 / TRU Concentrator for every CANDU
reactor.
PRIOR STUDENT WORK:
The
Ottensmeyer Plan originated in student work at the University of
Toronto circa 2010. The following notes are an edited version of
student work. There are various claims made by the students based on
references that should be confirmed before large sums of money are
committed to implementation of the process.
2.1.1 Fuel Bundle
Shearing CANDU nuclear fuel bundles are made from fuel pellets,
inserted into zircaloy cladded fuel rods and loaded into channels of
a cylindrical metal assembly (See Appendix B for diagram and
composition). Thus, the first step of reprocessing begins with the
removal of these long, narrow fuel rods from the bundle and
liberating the spent fuel. The proposed process achieves this by
mechanically shearing the rods into small segments, approximately 3cm
[6], and dissolving the exposed fuel in a hot nitric acid dissolver
solution.
Other alternatives for fuel liberation were considered,
such as chemical decladding, mechanical decladding and perforating
the cladding. Decladding proved uneconomical due to excess waste and
high losses, while perforating the cladding alone does not provide
enough exposed fuel surface area to dissolve it in a timely fashion
[6]. Sawing as an alternative to mechanical shearing was also
considered; however, it is less consistent and produces more metal
fines [7].
2.1.2 Dissolution The dissolver selected is a
countercurrent flow, multistage, rotary dissolver. After shearing,
pellets are fed into one end of the dissovler, while hot nitric acid
is fed into the other end at 4M and 95°C [6]. The
resulting solution, after dissolution of the spent fuel, is
approximately 300g/L (using U as a basis). The concentration and
temperature of nitric acid was found by analysis of calculated rate
data and experimental data (see Appendix C). While higher
concentrations of nitric acid yield higher rates of dissolution (75
minutes versus approximately 3 hours), it also introduces further
hazards with regards to acidity and corrosion. The dissolver stage
was not rate limiting in the process and thus the lower, less
hazardous acid concentration could be applied. As the dissolver drum
rotates, the pellets in the initial stage are transferred along its
length, while nitric acid flows counter-currently dissolving the fuel
within. The drum is rotated to propel the solids forward and moves in
a rocking motion to establish appropriate agitation for speedier
dissolution. It also allows for better mass transfer and more
efficient dissolution, as the least soluble fuel particles are
contacted with the strongest acid [7]. See Appendix E-1 for
schematic. At the last stage of the dissolver, the solid cladding is
ejected from the bottom, while the loaded nitric acid, containing
uranium nitrate, plutonium nitrate and other dissolved fission
products and actinides, flows out from the opposite end. The main
dissolution reactions (Rxn. 1 and 2) are the dissolution of uranium
dioxide (UO2) in nitric acid. Rxn. 2 is dominant when nitric acid
concentration is less than 10M [7]. The other actinides dissolution
reactions proceed similarly. UO2+ 4HNO3 = UO2(NO3)2+ 2NO2+ 2H2O
ΔHr°= -74.9 kJ/mol [Rx.1] 3UO2 + 8HNO3 =
3UO2(NO3)2 + 2NO + 4H2O ΔHr°= -367.6 kJ/mol
[Rx.2] During dissolution, NOx gases and radioactive iodine vapours
from the fuel are emitted and sent for gas treatment (see section
6.2) while the concentrated solution is sent for crystallization. The
undissolved cladding is rinsed, monitored for fissile material,
packaged and transferred to the solid waste storage area for
disposal.
This dissolver design is an update on more traditional
spent fuel dissolvers that involve placing pellets into perforated
baskets, followed by immersion in hot nitric acid. There are a number
of operating limitations associated with this method, including the
requirement of batch processing. Furthermore, the use of a highly
corrosive solvent, off-gas emissions, as well as the potential for
criticality, leads to challenges and limitations on the amount
processed per batch. The equipment also decreases any criticality
risk due to the processing of less mass in one location and its long,
narrow geometry [7].
2.1.3 Crystallization and Clarification The
dissolver solution contains both long lived actinides used for FNR
fuel and shorter-lived fission products. Before separation of
actinides from fission products, the majority of U can be
crystallized out of solution directly into Uranyl Nitrate Hexahydrate
(UNH), a yellow green crystal. Due to its high concentration of U in
solution (300g/L present as uranyl ions, UO22+), as well as its
relative insolubility, a decrease in solution temperature to 10-20°C
is capable of crystalizing approximately 70-80 wt% of the U [8].This
was determined based on solubility-temperature data, as well as
experimental results (see Appendix D). Co-crystallization of smaller
amounts of Pu(VI) and Np(VI) is possible and favourable, as this
would further diminish the amount of actinides requiring later
processing. The crystallization reaction proceeds as follows: UO22++
2NO3- + 6H2O = UO2(NO3)2•6H2O ΔHr°=
-20 kJ/mol [Rx.3] One issue with crystallization is the contamination
of U with fission products. Testing has shown that there is
negligible occlusion of fission products within the crystals, and
crystals can be washed to achieve a decontamination factor of about
100 for fission products (after 3 wash cycles) [8]. Washing is done
with cooled water in the last stages of a multistage crystallizer.
Although crystallization prior to extraction is not carried out in
conventional PUREX, this modified process uses it to reduce the
amount of aqueous and organic solution being processed. This addition
provides process and economic benefits (e.g. less process fluid), as
well as environmental benefits as the corresponding liquid waste
requiring treatment and disposal is reduced. While it may be possible
to achieve greater amounts of U crystallization with lower
temperatures (i.e. ~99% at -30°C [8]), liquid-liquid
extraction equipment (see Section: 2.1.4 Main Liquid-Liquid
Extraction and Stripping) would still be required to achieve
separation of the remaining actinides. The smaller temperature
reduction for approximately 70% U crystallization maximizes the
benefit of crystallization, without incurring unnecessary costs and
complexities. Before proceeding to the main extraction stage the
exiting dissolver solution must be clarified, also by centrifugation,
to remove any suspended particles that may interfere in extraction.
The collected particles are recycled for further dissolution. The
dissolver solution then proceeds to a mixing tank for acidity and
concentration adjustment, mainly to maintain the high concentration
of nitric acid (approximately 5-6M) required for high efficiency
separation in the main extraction stage [4]. The UNH crystals which
exit the crystallizer as a slurry, are gravity fed into a mixing tank
for further processing (see section 2.1.7 Metal Formation for more
details). The crystallizer selected is an annular, rotary,
screw-type, continuous crystallization device, with a cooling jacket
for temperature control. This design was selected due to its
continuous operation and its similarity in functioning to the other
screw-type equipment used in the overall process. The crystallizer
was designed to operate at an incline to aid in nucleation and growth
of crystals. This also facilitates easier removal of crystals without
damage. As the internal spiral blades rotate they dislodge the
crystals growing on the wall and move them upward to a centrifugal
basket for washing and filtration, while the remaining dissolver
solution flows downward. The rotary screw design, in a similar way to
the rotary dissolver, also provides compartments or stages that allow
for crystal washing to be done internally. See Appendix E-1 for
schematic. One operational disadvantage of the crystallizer design is
the potential for crystal accumulation and blockage of discharge
streams. This can be mitigated by appropriate operation. For example,
the screw rotation, dissolution feed and coolant feed can be adjusted
to restrict new crystal growth or break down crystal agglomerates.
Scrub acid may also be fed to decompose any crystal blockages [9]
(see Section 3. for more details).
2.1.4 Main Liquid-Liquid
Extraction and Stripping After crystallization, clarification and
concentration, the dissolver solution is ready for the main
liquid-liquid extraction and stripping. The solution is fed into the
middle of a multistage centrifugal contactor to begin the separation
of the remaining actinides from fission products. [4] M. Nakahara, Y.
Sani, Y. Koma, M. Kamiya, A. Shibata, T. Koizumi and T. Koyama,
"Separation of Actinide Elements by Solvent Extraction Using
Centrifugal Contactors in the NEXT Process," J Nucl Sci Technol,
vol. 44, no. 3, pp. 373-381, 2007. [5] J. D. Law, T. G. Garn, D. H.
Meikrantz and J. Warburton, "Pilot-Scale TRUEX Flowsheet Testing
For Separation of Actinides and Lanthanides from used Nuclear Fuel,"
Separation Science and Technology, vol. 45, pp. 1769-1775, 2010. [6]
H. M. Mineo, H. Isogai, Y. Morita and G. Uchiyama, "An
Investigation into Dissolution Rate of Spent Nuclear Fuel in Aqueous
Reprocessing," J. Nucl. Sci. Technol., vol. 41, no. 2, pp.
126-134, 2004. [7] R. Jubin, "Spent Fuel Reprocessing," in
Introduction to Nuclear Chemistry and Fuel Cycle Separations Course,
Consortium for Risk Evaluation With Stakeholder Participation,
Nashville, 2008. [8] T. Takata, Y. Koma, Sato, Koji, Kamiya,
Masayoshi, A. Shibata, K. Nomura, H. Ogino, Koyama, Tomozo and S.-i.
Aose, "Conceptual Design Study on Advanced Aqueous Reprocessing
System for Fast Reactor Fuel Cycle," J Nucl Sci Technol, vol.
41, no. 3, pp. 307-314, 2004. [9] K. Ohyama and K. Nomura,
"Development of Uranium Crystallization System in “NEXTâ€ン
Reprocessing
Process," in Proceedings of International Conference on Advanced
Nuclear Fuel Cycles and Systems, Boise, 2007. Â Appendix
D: Crystallization of Uranium in Nitric Acid Data The following graph
depicts the solubility of uranium in nitric acid, as a function of
uranium and nitric acid concentration, plus temperature. Moving from
right to left, the uranium concentration decreases with temperature
before reaching a minimum point, where water and nitric acid
crystallize. To avoid this co-crystallization, a crystallizer must
operate to the right of this point. The graph shows that as the
concentration of nitric acid increases, the minimum point shifts from
right to left and a higher UNH crystallization is possible. To
maximize crystallization, the feed uranium and nitric acid
concentration should be maximized as well [6]. Figure 3 Solubility
Curves of Uranyl Nitrate The above equilibrium data is supported by
experimental data. Crystallization tests at high uranium
concentration (300-600 g/l) and high nitric acid concentration (4-6M)
to simulate spent fuel dissolver solution have yielded 70-80% uranium
recovery at 10-20°C, and up to 95% at -10°C
[7]. The results for a run with 500g U/l and 5M nitric acid are
summarized below: Figure 4 Uranium and nitric acid concentration in
mother liquor (left) and recovery of uranium (right) Appendix E-1:
Equipment Diagrams The following appendix features schematic diagrams
of certain non-standard key equipment to aid in the visualization and
understanding of their functioning, as described in Section 2.1 of
the Process Description, and also as discussed in the non-key unit
sizing section. Figure 5 Fuel Bundle Shearer [8] Figure 6 Continuous
Rotary Dissolver [9] Figure 7 Rotary Crystallizer Crystallizer is
scaled down from a tested device, having a feed rate of 1380L/hr and
a residence time of 1 hour [6]. The dimensions of internal rotary
cylinder with the blades of the tested crystallizer device are 11
centimeters of internal diameter and 50 centimeters by length [2].
Since the amount of uranium crystals to be dealt with is much less,
the dimensions are rescaled by reducing the original volume of
0.00475m3 by half to 0.002375m3 in order to design for smaller
blades. Cost Table Appendix T: Works Cited for Appendices [1]
Advamacs, "Concentration Calculator," [Online]. Available:
http://www.trimen.pl/witek/calculators/stezenia.html. [Accessed
November 2012]. [2] D. P. Jackson, "NWMO Background Papers:
Technical Methods; Status of Nuclear Fuel Reprocessing, Partitioning,
and Transmutation," NWMO, 2003. [3] D. Hart and D. Lush, "THE
CHEMICAL TOXICITY POTENTIAL OF CANDU SPENT FUEL," NWMO, 2004.
[4] L. Johnson and J. Tait, "Source terms for 36Cl in the
assessment of used fuel disposal," Atomic Energy of Canada
Limited Technical Record, 1992. [5] J. Tait, I. Gauld and G. Wilkin,
"Derivation of Initial Radionuclide Inventories for the Saftey
Assessment of the Disposal of Used CANDU Fuel," Atomic Energy of
Canada Limited Report, 1989. [6] G. Faure, Principles and
Applications of Geochemistry, Upper Saddle River: Prentic Hall Inc.,
1998. [7] R. Hart and G. Morris, "Crystallization temperatures
of uranyl ntirate-nitric acid solutions," Prog. Nucl. Energy
III, p. 544, 1958. [8] T. Chikazawa, T. Kikuchi, A. Shibata, T.
Koyama and S. Homma, "Batch Crystallization of Uranyl Nitrate,"
J. Nucl. Sci. Technol., vol. 45, no. 6, pp. 582-587, 2008. [9] T.
Todd, "Spent Nuclear Fuel Reprocessing," in Nuclear
Regulatory Commission Seminar, Rockville, 2008. [10] R. Jubin, "Used
Fuel Reprocessing," in CRESP Nuclear Fuel Cycle Course, 2011.
[11] Rousselet-Robatel, "ROUSSELET ROBATEL MODEL LX MULTISTAGE
CENTRIFUGAL EXTRACTOR," [Online]. Available:
http://www.rousselet-robatel.com/products/pdfs/multistage-centrifugal-extractor-operatingprinciple.
pdf. [Accessed 2012]. [12] P. A. Haas, R. D. Arthur and S. W.B.,
"Development of Thermal Denitration to Prepare Uranium Oxide and
Mixed Oxides for Nuclear Fuel Fabrication," Oak Ridge National
Laboratory, Oak Ridge, 1981. [13] S. Jeong, S. Park, S. Honge, S.
C.S. and S. Park, "Electrolytic production of metallic uranium
from U3O8 in a 20kg batch scale reactor," J. Radioanal. Nucl.
Chem., vol. 268, no. 2, pp. 349-356, 2006. [14] T. Takata, Y. Koma,
Sato, Koji, Kamiya, Masayoshi, A. Shibata, K. Nomura, H. Ogino,
Koyama, Tomozo and S.-i. Aose, "Conceptual Design Study on
Advanced Aqueous Reprocessing System for Fast Reactor Fuel Cycle,"
J Nucl Sci Technol, vol. 41, no. 3, pp. 307-314, 2004. [15] R. Herbst
and M. Nilsson, "Standard and advanced separation: PUREX
processes for nuclear fuel reprocessing," in Advanced Separation
Techniques for Nuclear Fuel Reprocessing and Radioactive Waste
Treatment, 2011, pp. 141-173. This web page last updated June 21,
2021 A^4 B^2 I Tsors. Â
This web page last updated December 30, 2020